See what's going on in the chemistry¶
Debugging during runtime¶
ProDiMo writes more output and waits for interactive commands if you increase verbose_level in Parameter.in. Even better, leave verbose_level=0 there, but change chemistry_and_energybalance.f and uncomment a line like
if ((ix.eq.11).and.(iz.eq.10)) verbose=1which tells ProDiMo to increase verbose_level once a certain grid point is reached. The meaning of verbose is ...
verbose = -1 : no wait, only very little warnings
verbose = 0 : no wait, standard warnings
verbose = 1 : wait for return (or other command) after finishing one grid point
verbose = 2 : wait after every Newton-Raphson chemistry step
verbose = 3 : (can't remember)
verbose = 4 : check analytic against numerical chemical JacobianNow, if ProDiMo waits for input (verbose>0) you can just press ENTER to go to the next point. Or, you can give one of these commands
ENTER : next point
0...4 : change verbose level
Q : search for thermal bifurcations
character : go to chemical analysisMeaning of reaction types¶
The meaning of the chemical reaction types are described in the chemistry solver page (see chemistry_solver_design).
Analyze the chemistry¶
If ProDiMo waits for input (verbose>0) and you type a character, you are prompted for the name of a species for chemical analysis. Type 'q' if you want to return without analysis
input species to analyse ...
> H2O
gas temperature = 980.861597112949
H nuclei density = 1885537853.20952
chi = 112260.313525271
formation ... H2O
27 55 NN: H2 + OH --> H2O + H 2.11E-12 1.06E-06
918 10044 PH: H3O+ + PHOTON --> H2O + H+ 1.65E-02 8.64E-08
destruction ... H2O
806 4205 PH: H2O + PHOTON --> OH + H 1.80E+00 1.16E-06
915 10040 P3: H2O + PHOTON --> O + H2 6.40E-03 4.11E-09
particle density = 6.426E-07 concentration = 3.408E-16
total rate = 1.275E-22
input species to analyse ...
> qparticle density is in , concentration means , total rate is the sum of all rates , should be zero. The meaning of the reaction entries are as
27 55 NN: H2 + OH --> H2O + H 2.11E-12 1.06E-06
^^^^ ^^^^ ^^^^ ^^^^^^^^ ^^^^^^^^
running number type reaction rate coeff rate
reaction in [depends] [1/cm^3/s]
number UMISTThe rate is the rate coefficient multiplied by the particle densities of the reactants, except for ice desorption, which is more difficult.
Search for thermal bifurcations¶
If you press "Q" instead, ProDiMo varies the Tgas systematically between 20K and 10000K, calls the chemistry, calculates the net heating function Q(Tgas) and lists the results, before going to the next grid point. For thermal stability Q(Teq)=0 and Q'(Teq)<0 at the equilibrium gas temperature Teq=Tgas(Q=0). It is possible that there exists more than one root of Q(Tgas)=0, a physical non-uniqueness of the condition of gas energy balance, sometimes referred to as "thermal bifurcations". You can manually search for these bifurcations this way.
Save and analyse chemical rates¶
The switch
.true. ! chemanalysiswill ask ProDiMo to write a special output file chemanalysis.out. If ProDiMo is run with .true. ! time_chem_disk the output will have a postfix chemanalysis_xx.out for each time-step of the time-dependent chemistry model (i.e. in the same way as for the ProDiMo.out). This file contains all the required information such as chemical rates for each grid point, and can be analysed either with IDL scripts or prodimopy.
The chemanalysis data can be read by IDL routines (see below) or by using the tools included in , it also includes some notebooks on how to use it, see the notebooks folder of the git repository.
Analysing with prodimopy¶
If you work with prodimopy, we recommend to use in Parameter.in
.true. ! chemanalysis_fitsinstead of chemanalysis. In that case the output is written to a fits file, which results in a much smaller file size, and improves the performance of the analysis scripts.
Examples on how to analyze the chemistry and make some figure can be found in the chemanalysis example notebooks.
Analysing with IDL¶
Note: I think at the moment IDL cannot deal with the time-dependent chemanalysis (with prodimopy it can be done).
IDL> .compile ../idl/prodimo
IDL> .compile ../idl/chemanalyse
IDL> chemanalyse,'name',/readdatawhere name is the name of a species, for example
IDL>chemanalyse,'H2O' ,/readdataThe switch is threadsafe because the output starts with the grid position ix and iz and the IDL routines can read the output in any order.
The IDL routine will write on screen all the formation and destruction reactions for this species. It will also output an ascii files chemistry_reactions_name.txt listing the main formation and destruction reactions. The first column is an index. The indices are used in the ps output files named chemistry_analysis_name.ps, where again the name is the name of the species. The optional input /readdata is only necessary the time to create a xdr file, which is much faster to read for IDL if, for example, you ask for another species to be analysed (see example below).
Example of chemistry_reactions_H2O.txt
-----------------------------------------------------
Main formation and destruction reactions
species :H2O
Formation reactions
1 31 55 NN: H2 + OH -> H2O + H
2 169 421 NN: NH2 + NO -> N2 + H2O
3 173 426 NN: OH + OH -> H2O + O
4 583 2119 IN: H3O+ + Si -> SiH+ + H2O
5 942 3563 DR: H3O+ + e- -> H2O + H
6 999 4084 RA: H + OH -> H2O + PHOTON
7 1149 4557 CL: H + OH -> H2O + M
8 1183 10044 PH: H3O+ + PHOTON -> H2O + H+
9 1197 10273 NN: H2exc + OH -> H2O + H
10 1211 10288 DT: H2O# + dust -> H2O + dust
11 1213 10290 DP: H2O# + PHOTON -> H2O +
Destruction reactions
1 9 10 NN: H + H2O -> OH + H2
2 355 1191 IN: C+ + H2O -> HCO+ + H
3 573 2061 IN: H2O + Si+ -> SiOH+ + H
4 577 2067 IN: H2O + SiH+ -> Si + H3O+
5 633 2951 CE: H+ + H2O -> H2O+ + H
6 1050 4205 PH: H2O + PHOTON -> OH + H
7 1051 4206 PH: H2O + PHOTON -> H2O+ + e-
8 1210 10287 IC: H2O + dust -> H2O# + dust
-----------------------------------------------------As usual name# means the icy species.
The figures show the main and second formation and destruction reactions at each location in the disk where the numbers are the reaction indices found in the chemistry_reactions_name.txt
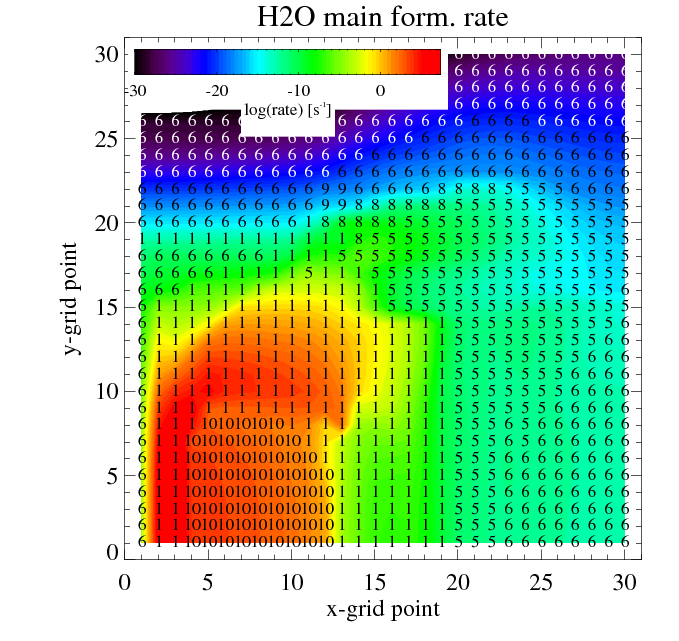
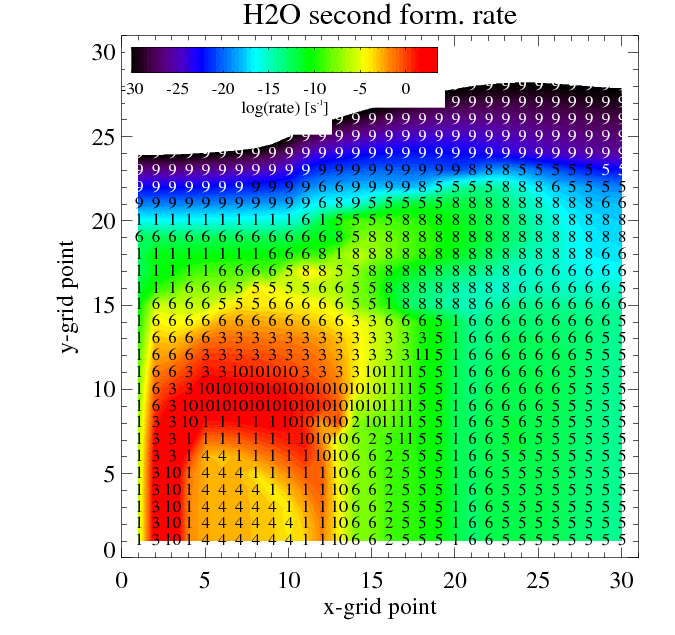 Filled contours: rate of the main (left) and second (right) most formation reaction. Numbers: index of the reaction
Filled contours: rate of the main (left) and second (right) most formation reaction. Numbers: index of the reaction
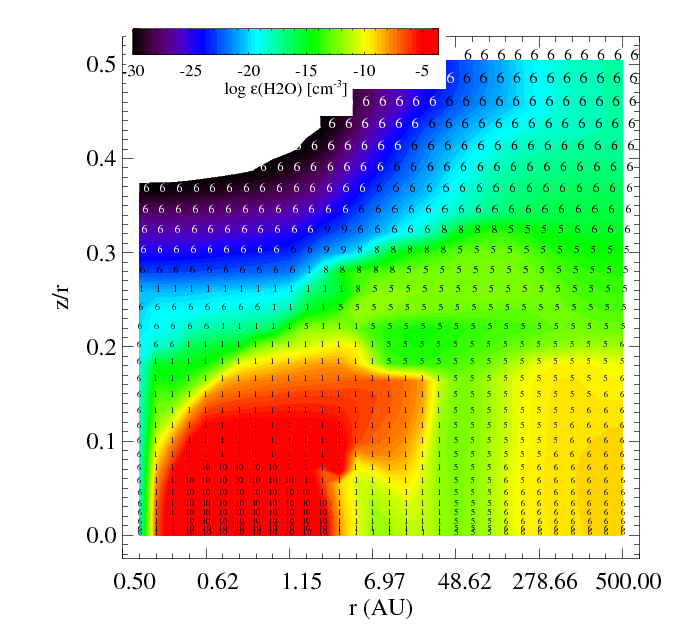 |
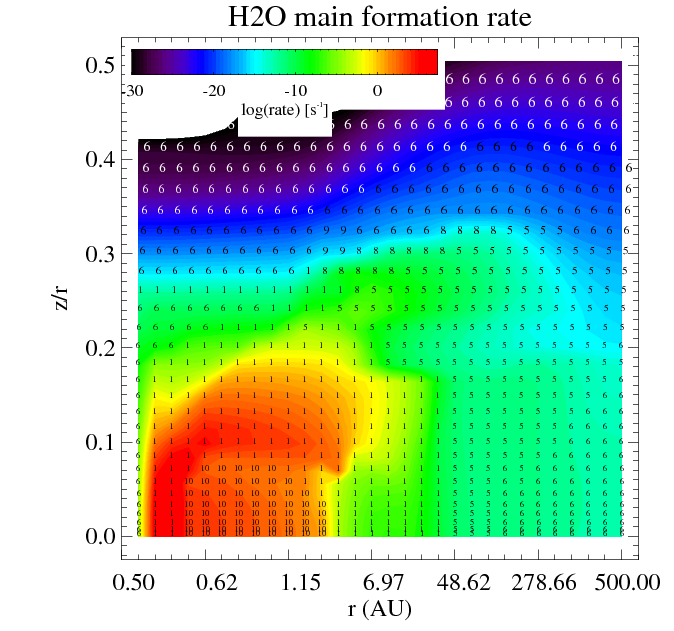 |
| Filled contours: water relative abundance epsilon. Numbers: index of the reaction | Filled contours: total formation rate. Numbers: index of the reaction |
Similar plots are made for the destruction reactions.
After chemanalysis.pro is finished producing the ps-file, the user can enter a manual point-by-point analysis, with similar output as described on the top of this page. Start by entering the point-coordinates ix,iz, then you can have a look at the most important formation and destruction rates of selected molecules one by one. Don't be surprised if the total formation and total destruction rates are not entirely equal, because in the chemanalysis-mode, only a few of the most relevant reaction rates are stored on file.
Do you want to analyse one point, manually? ix,iz=? (0,0 = exit)
: 70,39
grid point = 70 39
radius[AU],z/r = 151.18 0.1480
n<H>,nd[1/cm3] = 1.83E+06 7.26E-05
Tgas,Tdust[K] = 44.7594 58.4410
AV_rad,AV_ver = 2.01E-01 2.87E-04
abundant molecules: H2 H He O C Ne N CO Ar e- H2exc H+ Ne+ S+ C+ CH3+ C2 CH C2H
------------------------------------------------------------------------
nmol( HCO+)[1/cm3], concentration = 4.683E-03 2.553E-09
Total formation rate [1/cm3/s] = 7.8321741e-09
931 2321 IN: CH3+ + O -> HCO+ + H2 7.38E-09
1669 3919 IN: O + C2H2+ -> HCO+ + CH 2.07E-10
1 64 AD: CH + O -> HCO+ + e- 1.12E-10
1674 3926 IN: O + C3H3+ -> HCO+ + C2H2 5.95E-11
890 2221 IN: CH2+ + O -> HCO+ + H 4.36E-11
1113 2664 IN: H2 + CO+ -> HCO+ + H 2.85E-11
Total destruction rate [1/cm3/s] = 7.8774804e-09
646 1357 DR: HCO+ + e- -> CO + H 6.61E-09
830 2134 IN: C + HCO+ -> CO + CH+ 1.26E-09
2200 5957 PH: HCO+ + PHOTON -> CO+ + H 7.31E-12
1730 4032 IN: OH + HCO+ -> HCO2+ + H 1.09E-13
1174 2777 IN: H2O + HCO+ -> CO + H3O+ 2.51E-14
1316 3137 IN: HCO+ + C3H2 -> C3H3+ + CO 2.17E-14
------------------------------------------------------------------------
: CH3+
nmol( CH3+)[1/cm3], concentration = 3.379E-02 1.842E-08
Total formation rate [1/cm3/s] = 1.3816863e-07
1110 2658 IN: H2 + CH2+ -> CH3+ + H 1.38E-07
1279 3069 IN: H + CH4+ -> CH3+ + H2 1.05E-10
2161 5876 PH: CH3 + PHOTON -> CH3+ + e- 3.17E-11
1676 3957 IN: O + CH4+ -> OH + CH3+ 2.73E-11
2332 10029 PH: CH5+ + PHOTON -> CH3+ + H2 3.35E-12
1671 3921 IN: O + C2H4+ -> CH3+ + HCO 1.28E-12
Total destruction rate [1/cm3/s] = 1.3857571e-07
2159 5874 PH: CH3+ + PHOTON -> CH2+ + H 2.97E-08
2158 5873 PH: CH3+ + PHOTON -> CH+ + H2 2.97E-08
586 1184 DR: CH3+ + e- -> CH + H2 2.70E-08
587 1185 DR: CH3+ + e- -> CH + H H 2.29E-08
585 1183 DR: CH3+ + e- -> CH2 + H 1.07E-08
824 2124 IN: C + CH3+ -> C2H+ + H2 9.92E-09
------------------------------------------------------------------------
: <-- just hit RETURN here to exit this loop
Do you want to analyse one point, manually? ix,iz=? (0,0 = exit)
: 0,0
% Stop encountered: CHEMANALYSE 1407 /home/star/pw31/ProDiMo/idl/chemanalyse.proAn other option is
IDL> reaction_network,8,25where 8 and 25 are the x and z grid index. The IDL routine will list on the screen the main formation and destruction reactions for all the species at the grid location (x,z).
Make species network
1 1st : 622 species :He nparents = 2 :H He+ -> He H+
1 1st : 1095 species :He nchildren = 1 :He CRP -> He+ e-
2 1st : 1095 species :He+ nparents = 2 :He CRP -> He+ e-
2 1st : 622 species :He+ nchildren = 2 :H He+ -> He H+
3 1st : 971 species :C nparents = 1 :C+ e- -> C PHOTON
3 1st : 1021 species :C nchildren = 1 :C PHOTON -> C+ e-
4 1st : 1021 species :C+ nparents = 2 :C PHOTON -> C+ e-
.......