Grain surface chemistry¶
The treatment of surface chemistry is explained below. The H2 formation via surface chemistry is described in Thi et al.(2020).
"New" Surface chemistry ProDiMo version >= 2.0¶
A simple surface chemistry network has been added. The implementation uses the rate equation approach of Hagesawa et al. (1992) and Hagesawa et al. (1993) with modifications to include cosmic-ray enhanced surface diffusion, direct evaporation of products resulting from exothermic reactions, photodissociation of surface species, tunnelling for all species for diffusion and to overcome reaction barriers. A recent description of the method can be found in Reboussin et al. (2014) and Garrod & Herbst (2006). The grain-surface chemistry model in ProDiMo considers both the Langmuir-Hinselwood and Eley-Rideal mechanisms (as in the previous version), which might be important for water formation (Hollenbach et al. 2009).
The aim of the surface chemistry so far is to estimate abundances of relatively simple species in the photodesorption layers.
- A statistically well-mixed ice mantle. We do not differentiate between surface and the bulk of the ice within a few layers.
- choice of the number of reactive ice layers: no diffusion between the reactive and inactive layers
- The code is flexible in the sense that most uncertain parameters are treated as input parameters whose values can be varied by the users:
- spherical grains, 1.5e15 sites for a one micron radius grain, the number of sites varie with the grain radius. The value is hardcoded.
- rate equation approach
- the rate coefficients take into account (a flag can be used to disable it) the competition between diffusion and reaction (c.f. Garrod private comm.)
- Eley-Rideal and Hinselwood type of reactions (Tgas and Tdust can be different)
-
, with a free-parameter (0.3 to 0.5). Special cases exit for species with measured Ediff to use a unique value of k for all the species
.true. ! unique_fbinding 0.5 ! fbinding -
adsorption energy from various sources: discrepancies between different sources can be large
- H2 formation via Jura model, Groningen's model (Stephanie Casaux), or via surface chemistry
- the surface chemistry network is limited to small species up to methanol. No spin-dependent reactions nor deuterium reactions
- photodesoprtion: single UV field value or via UV photodesorption cross-section
- reactive desorption whose efficiency depends on a free parameters (Rice-Ramsperger-Kassel theory), default =0.01 (Garrod+2007)
- cosmic-ray induced spot desorption and enhanced surface reaction rates
- tunneling (diffusion and over reaction barriers) for all species scaled with the species mass. We consider rectangular barriers.
- the surface-network can be used in conjunction with any current gas-phase warm and cold gas network (UMIST. OSU, KIDA) + private neutral-neutral and 3-body reaction network
- the gas-phase photo reaction rates can be treated using UV radiative-transfer and UV-destuction cross-sections or via scaled interstellar UV field
- PAH freeze-out, PAH are considered not reactive once frozen-out (only one PAH size is considered)
- grain charging is treated explicitly with no numerical restriction on the amount of charges per grain apart from grain physics
- photodestruction of surface species via equivalent ISM UV field method
- X-ray gas-phase chemistry can enhance the amount of available atomic hydrogen atoms, making surface reactions very efficient
- no X-ray desorption of photo-induced surface reactions
- hot atom (a parameter between 0 and 1) for the efficiency of using the adsorption energy (from the adsorption potential) to overcome part of the energy barrier for endothermic Eley-Rideal reactions A new treatment of the rate coefficients has been implemented. The surface chemistry option is in the beta phase and should not be used for research purpose without the help of the code developers. In particular, the network is incomplete and many important processes (such as the formation of H2 at high dust temperature has not been implemented yet). Please contact W.-F. Thi if you intent to test/use it. The routines still have to pass tests (see Semenov et al. (2010)
The Elements.in, Specie.in, Molecular_Clouds_Inputs.in and Parameter.in files to run a cloud model can be found in ProDiMo_path/input_examples/surface_chemistry.
In the molecular cloud mode, the parameter file contains in addition to the standard input the following switches:
.true. ! chemanalysis
.true. ! mc_only : compute Molecular Cloud abundances only
.true. ! mc_grid
.true. ! surface_chemistry
0.01 ! evap_fac
.true. ! photodesorption
.true. ! DVODE
.true. ! tunnellingThe chemanalysis.pro routines have been updated to analyze Molecular Cloud runs. The value for evap_fac controls the amount of chemical products that are back to the gas-phase because the reactions are exothermic (Garrod 2007). If tunneling through energy barriers is important than the switch should be on (default=on, so the switch is by default not needed). Minissale et al. (2013) showed that tunneling for species as heavy as oxygen can be important. Goumans & Andersson (2010) also studied theoretically that for the O + CO reaction tunnelling strongly increases the reaction rates at low temperature (10–20 K).
The solver DVODE has been adapted to work smoothly with surface reactions networks, please use it for Molecular Cloud models. An alternative solver from the Numerical Recipes book (.true. ! STIFBS) can be used from Revision 2265. Both solvers use separate matrix solvers and routines, thus they allow the numerical noises to be checked. The main switch is
.true. ! surface_chemistry : activate surface chemistryin Parameter.in. For the moment a very restricted network for the main species are available.
The Molecular_Cloud_Input.in reads
-----------------------------------------------------------------
1 ! nmodels : number of molecular cloud models
0 ! verbose_level : how much output? (-1...4)
---------------------------------------------------------------
*** Run a default molecular cloud model with surface chemistry
2e4 ! nH_mc : molecular cloud number density in cm^-3
15. ! Td_mc : dust temperature in K
15. ! Tg_mc : gas temperature in K initial guess (in case of thermal balance calculations)
1.0 ! CHI1_mc : UV field in band 1
1.0 ! CHI2_mc : Uv field in band 2
0.1 ! a1_mc : grain mean radius in micron
0.01 ! dust_to_gas_mc
3.0 ! rho_gr_mc
0.2 ! vturb_mc [km/s]
10.0 ! Av_mc : extinction to the edge of the cloud
test_mc1_surface_chemistry.out ! model_filename
25 ! N_age_mc : number of age bins (the ages are give in the next line in years)
10. 1e2 2e2 3e2 4e2 5e2 6e2 7e2 8e2 9e2 1e3 5e3 1e4 1e5 1e6 1.1e6
1.2e6 1.3e6 1.4e6 1.5e6 1.6e6 1.7e6 1.8e6 1.9e6 2.0e6
---
.false. ! mc_steady_state
.false. ! mc_thermal_balance
.true. ! end : obligatory at the end of each model descriptionthe surface chemistry results can be plotted with the dedicated routine in IDL/ analyse_time_dependent_clouds_surface_chem.pro
IDL> analyse_time_dependent_clouds,'test_mc1_surface_chemistry.out','surf_chem.ps',10.where 10. is the minimum age for the plot. Other IDL options are
,tmax=1e6 if you want to plot up to 1e6 yrs.
,ages='filename.txt' to specify the name of the file for the agesExamples of plots generated by the IDL script are given below
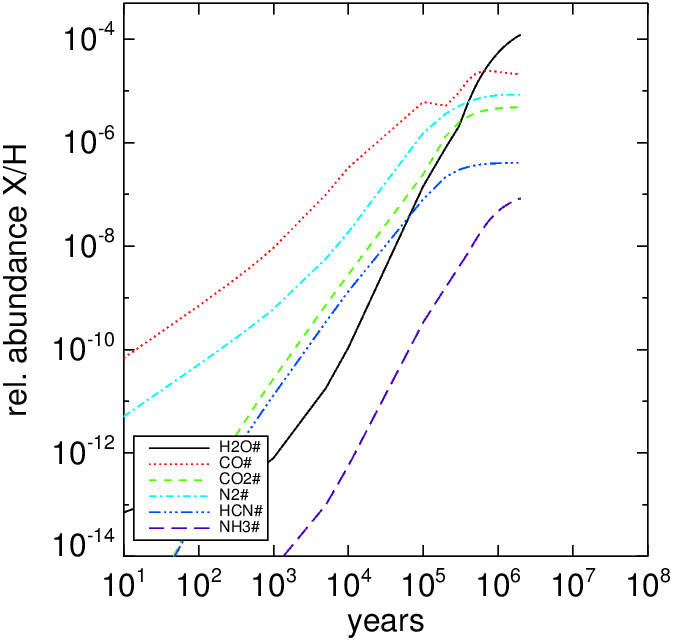
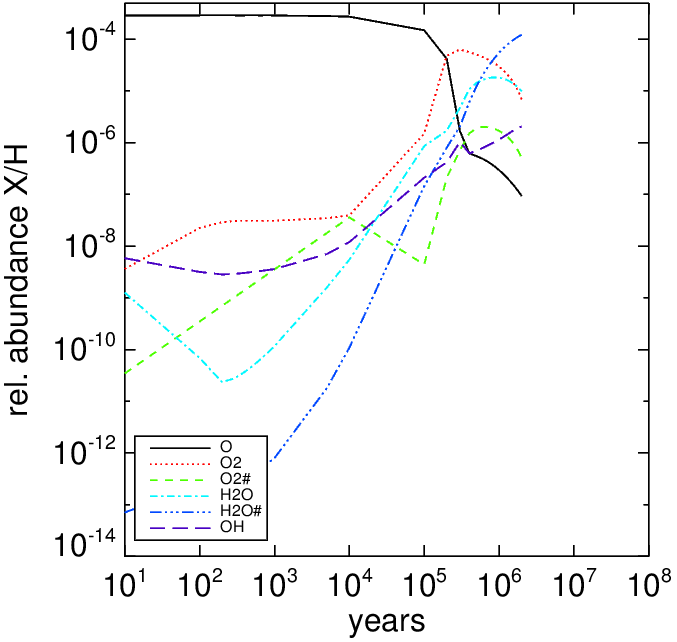 The left panel show the time-dependent abundance of the ice on grains for a molecule cloud model with , , and (All gas species were in atomic form except for H2). The right panel show the main oxygen-carriers for the same molecule cloud.
The left panel show the time-dependent abundance of the ice on grains for a molecule cloud model with , , and (All gas species were in atomic form except for H2). The right panel show the main oxygen-carriers for the same molecule cloud.
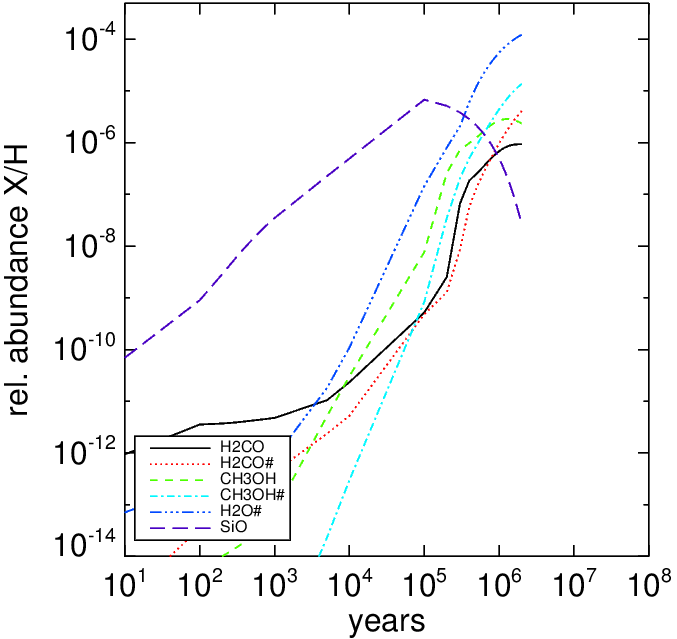 The left panel show the time-dependent abundance for formaldehyde and methanol. The right panel shows the abundance of N-species.
The left panel show the time-dependent abundance for formaldehyde and methanol. The right panel shows the abundance of N-species.
Individual formation and destruction processes can be studied using the chemanalyse.pro routine.
IDL> MC_chemanalyse,'H2O#','test_mc1_surface_chemistry.out',/readdataThe read data option has to be used if the MC_chemanalyse routine is used for the first time after the run. A sample of screen output:
Species :H2O#
All the formation reactions : 5
1575 10287 IC: H2O + dust -> H2O# + dust 1.45188e-13
1696 13203 GG: OH# + H -> H2O# + 4.67784e-12
1932 13781 S3: H# + OH# -> H2O# + 1.82054e+08
1968 13811 S3: H2# + OH# -> H2O# + H# 1.79547e-11
2060 13924 S3: H2O2# + H# -> H2O# + OH# 3.67240
All the destruction reactions : 5
1576 10288 DT: H2O# + dust -> H2O + dust 0.00000
1577 10289 DC: H2O# + CRP -> H2O + 1.05097e-42
1578 10290 DP: H2O# + PHOTON -> H2O + 1.09168e-16
2017 13859 S6: H2O# + PHOTON -> OH# + H# 2.44256e-17
2036 13897 S5: H2O# + CRP -> OH# + H# 2.14812e-31
---------------------
.....
---------------------
time= 1.50000e+06 yr
Species :H2O#
----------------------------------
Selection of the majors rates
All the formation reactions : 5
1575 10287 IC: H2O + dust -> H2O# + dust 1.45188e-13
1696 13203 GG: OH# + H -> H2O# + 4.67784e-12
1932 13781 S3: H# + OH# -> H2O# + 1.82054e+08
1968 13811 S3: H2# + OH# -> H2O# + H# 1.79547e-11
2060 13924 S3: H2O2# + H# -> H2O# + OH# 3.67240
All the destruction reactions : 1
2017 13859 S6: H2O# + PHOTON -> OH# + H# 2.44256e-17
abundance= 9.39031e-05
main creation reaction numbers : 1575 2060
main destruction reactions numbers : 2017
---------------------The entire network (all species at all ages) can be generated (on screen with the command): IDL> mc_reaction_network
The surface chemistry network also in the disk modeling mode. In Parameter.in, the following lines describe the way to call the surface chemistry routines:
.true. ! chemanalysis
.false. ! mc_only : compute Molecular Cloud abundances only
.false. ! mc_grid
.true. ! surface_chemistry
0.01 ! evap_fac
.true. ! photodesorption
.false. ! DVODE
.true. ! tunnelingIn this case the DVODE solver should not be used (not tested in parallel mode). A coupled of results of a HerbigAe disk model with and without surface chemistry. Notice that in the H2 formation in the surface chemistry mode do not have H2 formation on hot grains due to chemisorption sites, hence the lower H2 pure-rotational line fluxes (See figures below). Only physisorbed sites are modeled for the moment. The water line fluxes can be significantly enhanced. Notice the change in the location of the emission.
From Revision 09bad91e, it is possible to use the sticking coefficients published by Chaabouni et al. 2012 (A&A 538, 128 [http://adsabs.harvard.edu/abs/2012A%26A...538A.128C]) . In Parameter.in,
.true. ! Chaabouni_stickThe coefficients are those used to fit the Buch & Zhang 1991 simulations (ApJ, 402, 585). More recent simulations contradict the work of Buch & Zhang (Veeraghattam et al. 2014, ApJ 790, 4 [http://adsabs.harvard.edu/abs/2014ApJ...790....4V]). The main reason stems from the fact that Buch & Zhang used a small water cluster in their model. Chaabouni et al. formula gives a very low H sticking probability at high temperature for physisorption.
The main effect of surface chemistry is hydrogenation. Atomic oxygen is converted rapidly into water and CO into CH3OH. The efficiency of this conversion is at the heart of the discussions between surface chemistry researchers. Different processes (adsorprtion/desorption/diffusion at the surface/diffusion in the bulk/H-tunnelling,...) concur but the exact formulation is still a matter of debates in the community. Therefore the results from a model that includes surface chemistry should be taken will care. What is certain is that surface chemistry has a definitive effects for the abundance both in the gas and in the solid phase for a couple of important species such as methanol or water. Below are shown an example of the effects of including surface chemistry for the abundance and fluxes of H2 and H2O.
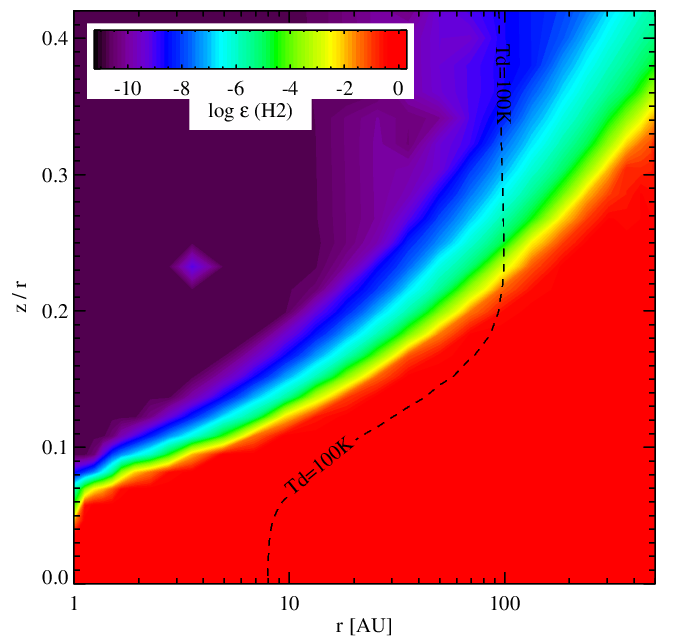
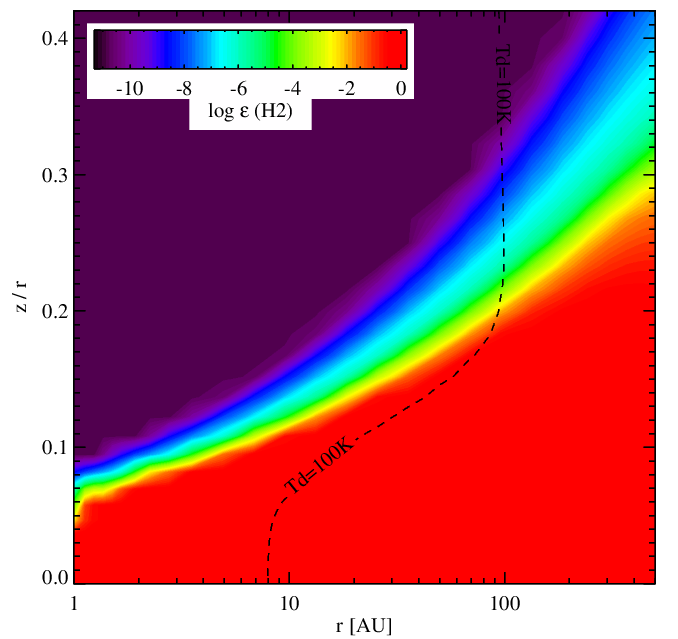 H2 abundance without (left panel) and with (right panel) surface chemistry. In the surface chemistry mode, the high-dust temperature method for forming H2 (H-chemisorbed sites) is disable for the moment. Therefore the H2 formation in disk surfaces is not as efficient.
H2 abundance without (left panel) and with (right panel) surface chemistry. In the surface chemistry mode, the high-dust temperature method for forming H2 (H-chemisorbed sites) is disable for the moment. Therefore the H2 formation in disk surfaces is not as efficient.
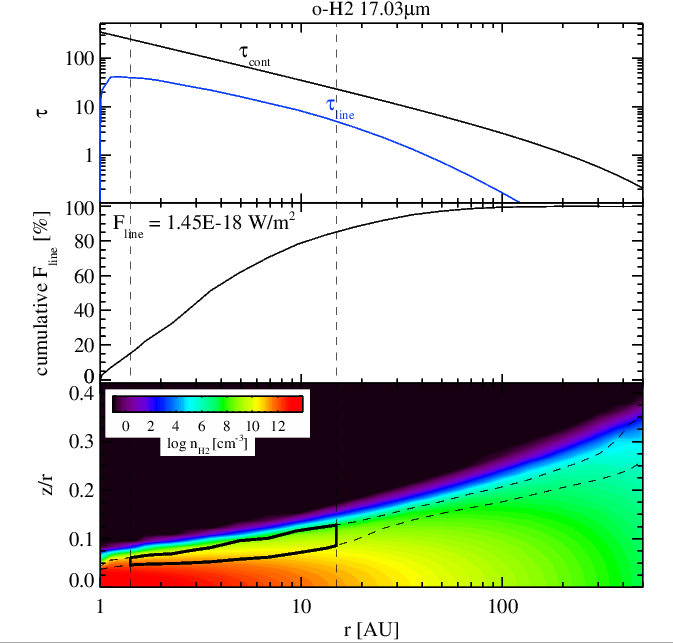
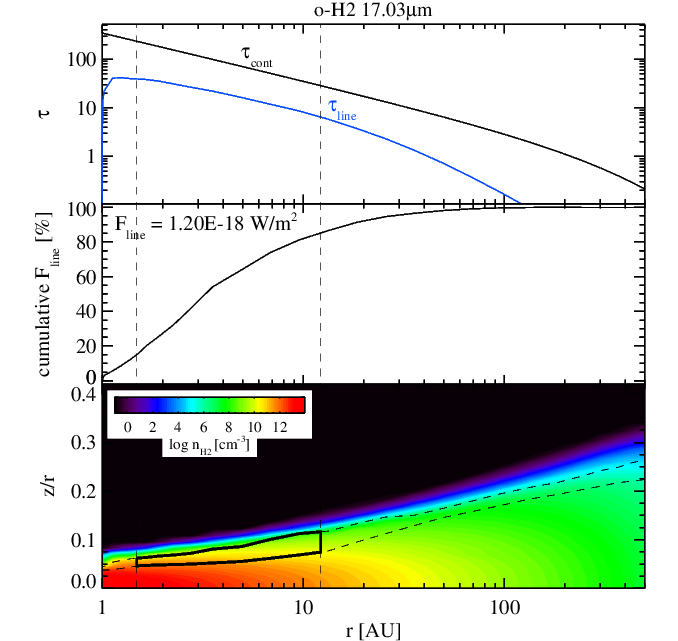 H2 17 micron emission without (left panel) and with (right panel) surface chemistry
H2 17 micron emission without (left panel) and with (right panel) surface chemistry
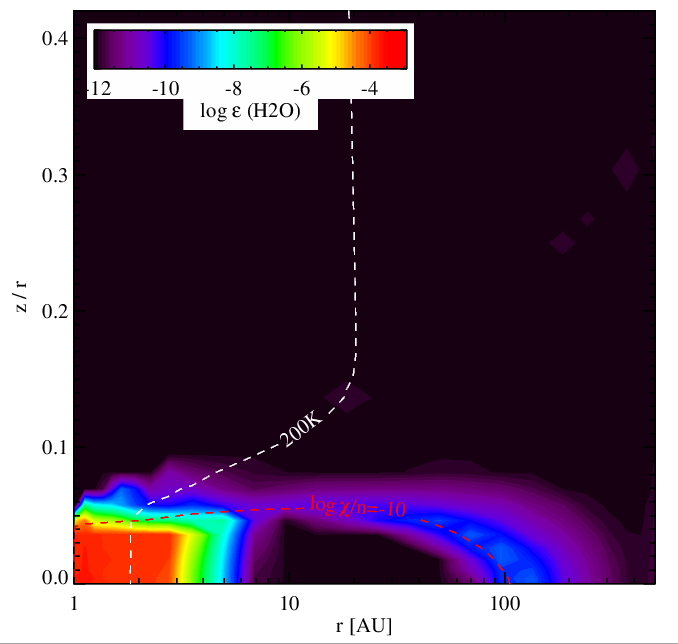
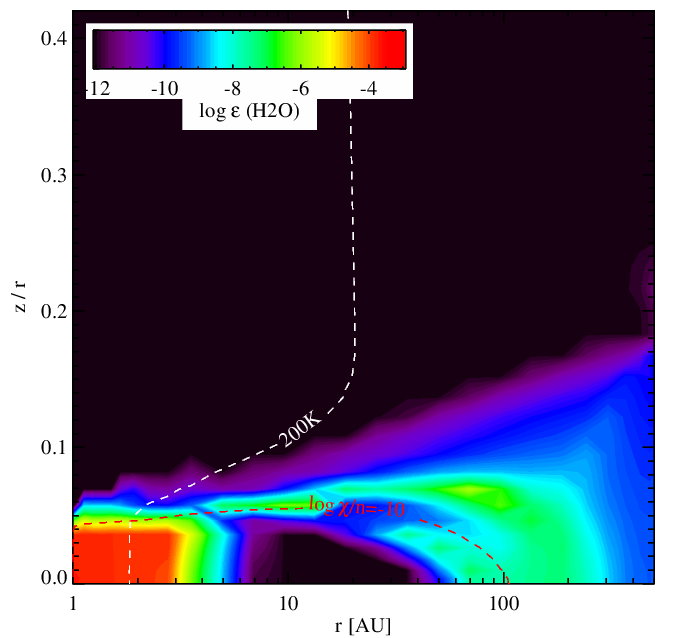 Water vapour abundance without (left panel) and with (right panel) surface chemistry
Water vapour abundance without (left panel) and with (right panel) surface chemistry
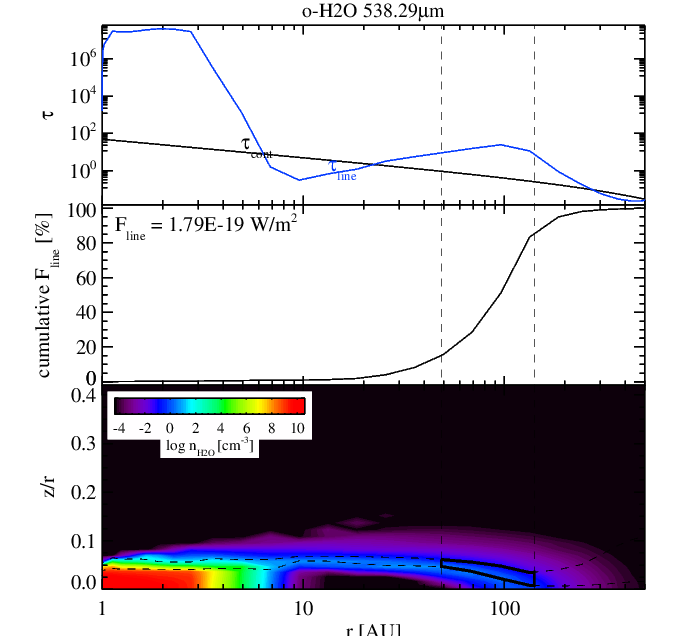
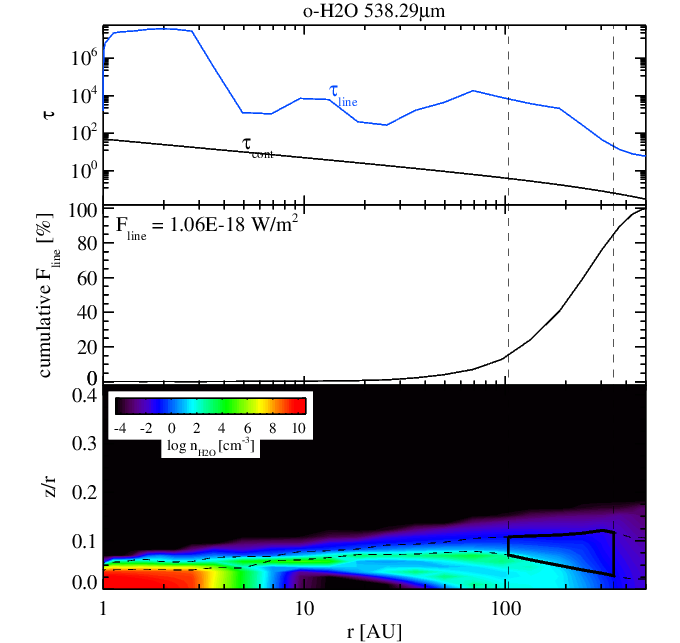 Water para fundamental line flux without (left panel) and without (right panel) surface chemistry
Water para fundamental line flux without (left panel) and without (right panel) surface chemistry
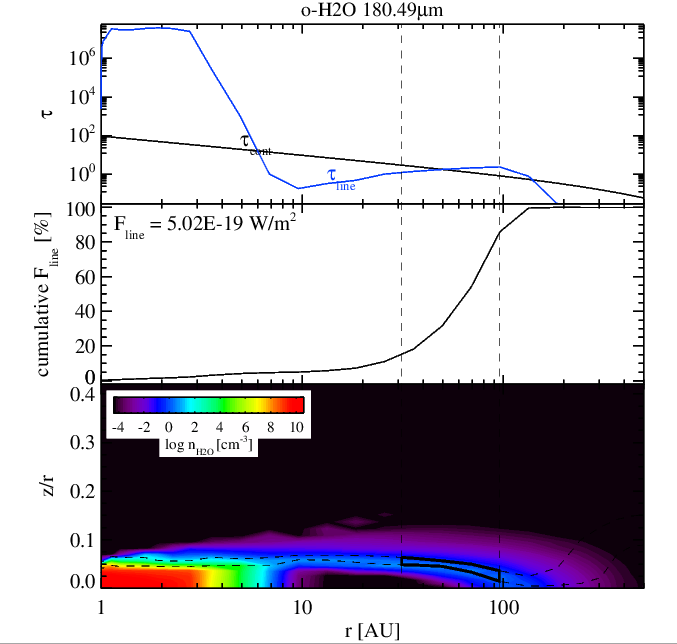
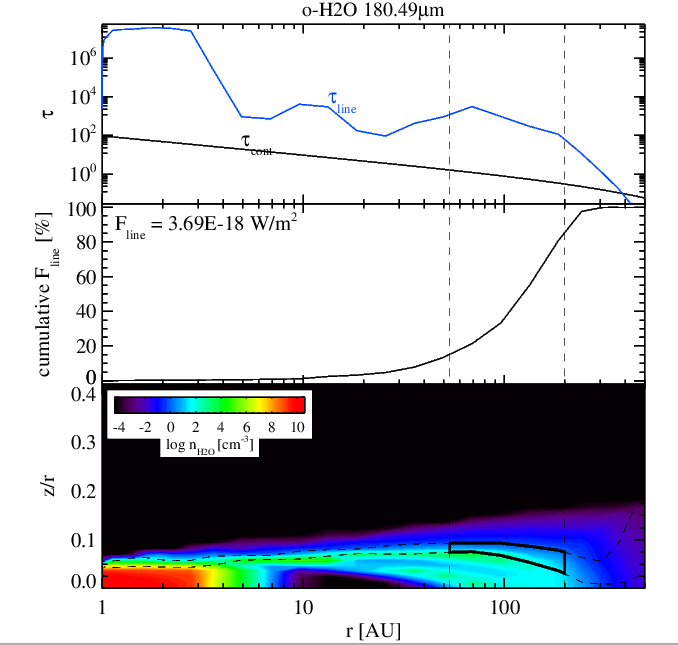 Water 180 micron line without (left panel) and with (right panel) surface chemistry
Water 180 micron line without (left panel) and with (right panel) surface chemistry
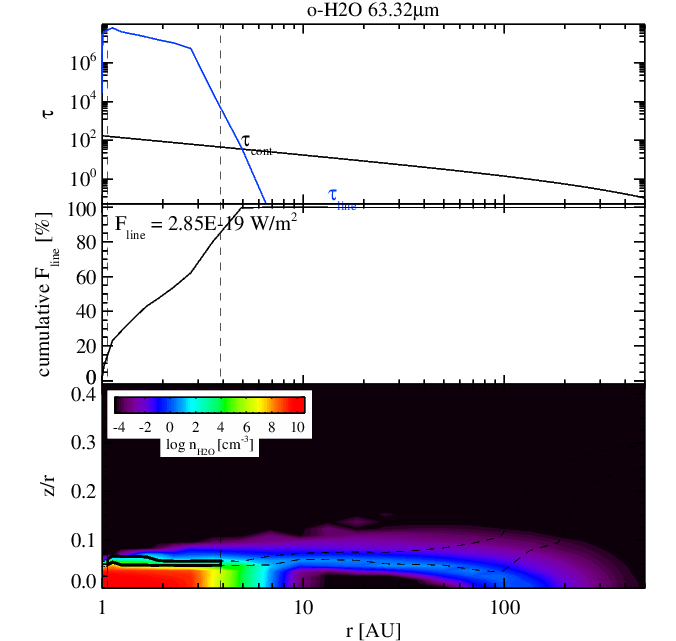
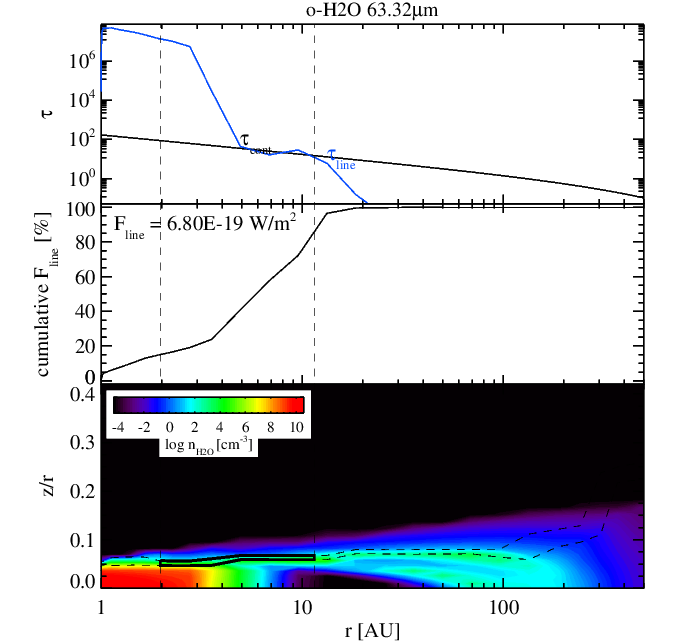 Water 63 micron line without (left panel) and with (right panel) surface chemistry
Water 63 micron line without (left panel) and with (right panel) surface chemistry
As of 15/10/2015
After discussions with surface experimental experts, it seems that the question of diffusion tunnelling is not solved yet. Thermal diffusion between shallow sites can also explain that the diffusion still occurs at T<10K. Independently of the reason for the low dust temperature hydrogen atom diffusion, this experiment effect has too be account for in the model. Therefore, it is still recommended to use the tunnelling switched on.
"Old" Surface chemistry (ProDiMo version < 2.0)¶
At this stage, the only reaction mechanism for surface chemistry included in ProDiMo is the Eley-Rideal mechanism. This means prompt reaction of a surface-bound atom/molecule with an incoming atom/molecule from the gas phase. The second mechanism (Langmuir-Hinshelwood) involving diffusion/tunneling on the grain surface is not yet taken into account since it requires treating the H2 surface formation consistently within the chemical network.
Examples for Eley-Rideal surface reactions are
O# + H -> OH#
OH# + H -> H2O#The corresponding entry in the Reactions.in file should read
3002 O# H OH# 4.95E-19 -0.38 0.0000
3003 OH# H H2O# 2.63E-18 -5.22 0.0000The calculation of the rates and Jacobians in explained in the appendix A of the paper Kamp et al. (2013).