How to create a simple mid-IR line spectrum based on the line fluxes computed with escape probabilities?¶
By default, ProDiMo calculates all line fluxes with escape probability and outputs them into the file "FlineEstimates.out". In the src_develop folder, you will find a simple FORTRAN program that creates a high-resolution wavelength grid, puts all line fluxes on their best-maching wavelength points, and then convolves the spectrum to a given spectral resolution, for example R=2500. This "escape probability spectrum" can be used, e.g., for a rough comparison to a continuum-subtracted JWST spectrum. In order to create the output files that can later be visualised, run "eprospec" in your ProDiMo run-folder like this:
> cd src_develop
> gfortran -fdefault-real-8 -fdefault-double-8 eprospec.f -o eprospec
> cd MyRunFolder
> eprospecwhich will create output files like these:
eprospec_C2H2_H.out
eprospec_CH4_H.out
eprospec_CO.out
eprospec_CO2_H.out
eprospec_H2.out
eprospec_H2O.out
eprospec_HCN_H.out
eprospec_NH3_H.out
eprospec_OH.out
eprospec_all.out
eprospec_ions.outThe output will depend on the molecules and lines included in your ProDiMo simulation. If you want additional molecules and different selection of lines, use a 'LineSelection.in' in your ProDiMo model. If you want a different wavelength range or different R, modify eprospec.f.
You can visualise the eprospec*.out yourself, or use this python code
> cd MyRunFolder
> python2 ~/ProDiMo/python/Plot_eprospec.py
> evince eprospec.pdf &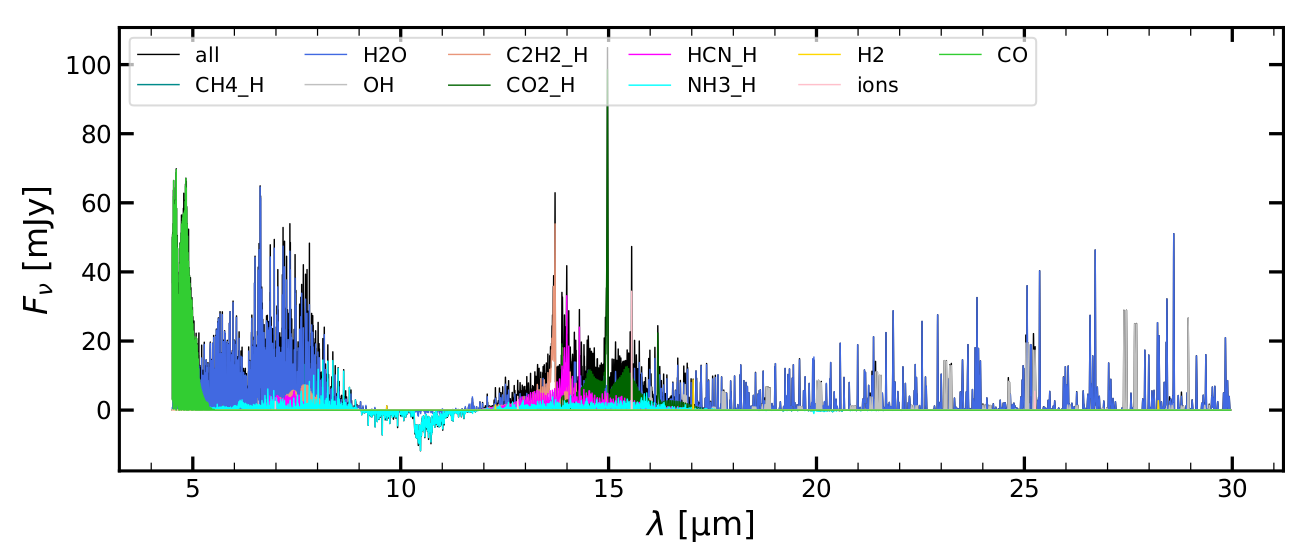
The screen output of Plot_eprospec.py lists some interesting integrated and line peak fluxes:
wavelength range [mic]: 4.9 8.0
molecules Fint[mJy*mic] Fpeak[mJy]
CH4_H 2.774906 17.190000
H2O 20.127571 64.560000
OH 0.000014 0.001479
C2H2_H 1.731339 7.269000
CO2_H 0.000000 0.000000
HCN_H 1.825778 5.523000
NH3_H 2.166184 10.080000
H2 0.000738 0.204600
ions 0.007412 2.492000
CO 1.842529 40.900000
wavelength range [mic]: 13.0 17.7
molecules Fint[mJy*mic] Fpeak[mJy]
CH4_H 0.000000 0.000000
H2O 5.321269 14.950000
OH 0.443380 7.417000
C2H2_H 9.818661 53.890000
CO2_H 9.211376 98.530000
HCN_H 6.897557 33.100000
NH3_H 3.380544 4.953000
H2 0.063390 8.735000
ions 0.227837 34.400000
CO 0.000000 0.000000The example spectrum was created using the examples/StandardObjects/TTauri_new model which has settle_method=3, a_settle=0.001, and newUVphoto=3.
How to create a proper mid-IR line spectrum with FLiTs, based on the disk shape, dust opacities and line populations calculated by ProDiMo?¶
First, you need to download and compile FLiTs, the Fast Line Tracer by Michiel Min, see wiki FLiTs for a few details. It is important that you pass the multi=true argument when compiling to make it openmp-parallel:
> cd FLiTs
> make multi=trueNext, you need to run ProDiMo with the .true. ! FLiTs flag in your Parameter.in. This will create a number of "*.Lambda.dat" files and the main exchange file "ProDiMoForFLiTs.fits" or "ProDiMoForFLiTs.fits.gz" or "ProDiMoForFLiTs.dat".
Now you are all set to run your FLiTs simulations. In order to automatise, I use a csh-script called run.FLiTsQuick. To prepare running that script, we need to compile a fortran program that reads the output from FLiTs and then convolves the FLiTs spectrum to a given spectral resolution.
> cd src_develop
> gfortran -fdefault-real-8 -fdefault-double-8 flitsspec.f -o flitsspecTo further prepare running the script, you need the following input files for FLiTs: 'H2O_quick.in', 'OH_quick.in', 'C2H2_quick.in', 'HCN'_quick.in, 'CO2_quick.in', 'NH3_quick.in', 'CH4_quick.in', 'CO_quick.in', 'ions_quick.in', 'H2_quick.in', 'all_quick.in', and 'SO2_quick.in'. You can extract these files from this tar-file inputFLITS.tgz. You need to put these files in a directory as specified in run.FLiTsQuick, in my case it's ${HOME}/FLiTs/input.
Now you can run the script like this:
> cd MyRunFolder
> ./run.FLiTsQuick > FLiTs.log &This takes about 30min on my Linux-PC. Once successfully run, you will find the following files in your MyRunFolder:
specFLiTs_C2H2.out
specFLiTs_CH4.out
specFLiTs_CO.out
specFLiTs_CO2.out
specFLiTs_H2.out
specFLiTs_H2O.out
specFLiTs_HCN.out
specFLiTs_NH3.out
specFLiTs_OH.out
specFLiTs_ions.out
specFLiTs_all.out
specFLiTs_conv_C2H2.out
specFLiTs_conv_CH4.out
specFLiTs_conv_CO.out
specFLiTs_conv_CO2.out
specFLiTs_conv_H2.out
specFLiTs_conv_H2O.out
specFLiTs_conv_HCN.out
specFLiTs_conv_NH3.out
specFLiTs_conv_OH.out
specFLiTs_conv_ions.out
specFLiTs_conv_all.outThe specFLiTs_*.out are the FLiTs output spectra, and the specFLiTs_conv_*.out are the convolved spectra generated by flitsspec. Now we can visualise the latter, in comparison to the eprospec_*.out
> python2 ~/ProDiMo/python/Plot_eprospec_FLiTspec.py
> evince eprospec_FLiTspec.pdf &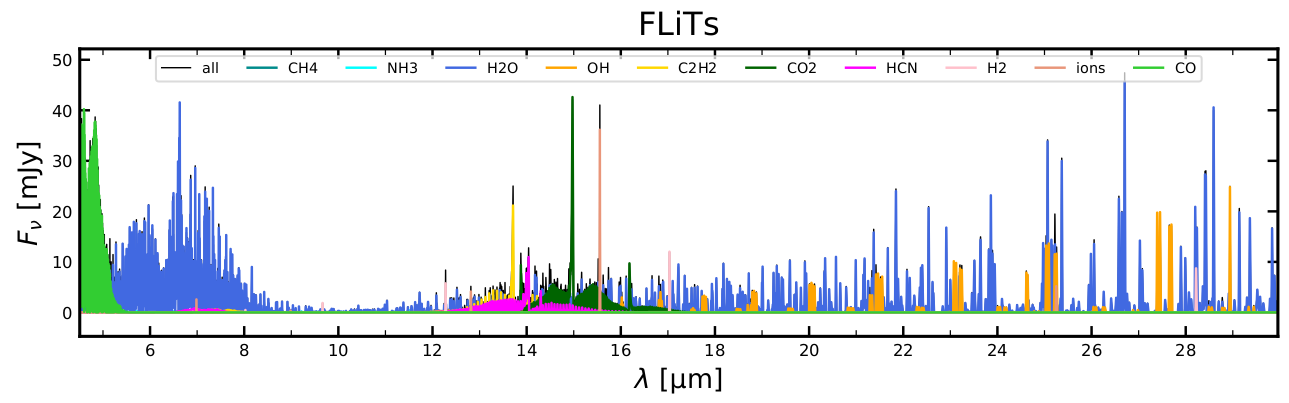
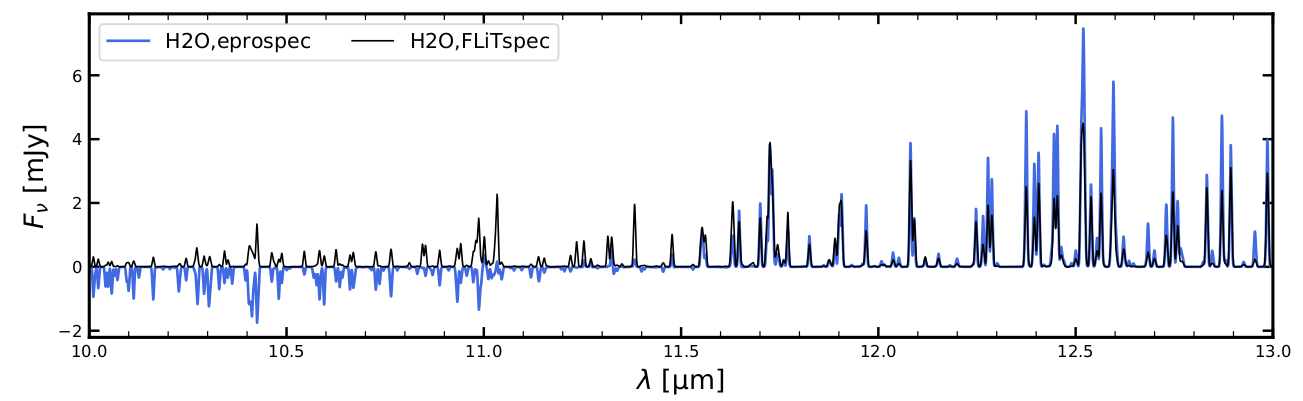
You will see some quite significant differences between the escape-probability-spectrum and the FLiTs-spectrum when you study the various eprospec_FLiTspec.pdf pages, for example the peak fluxes are almost a factor 2 lower, the FLiTs simulation does not confirm the NH3 absorption features as predicted by escape probability, etc. Some of the molecular emission lines are quite nicely predicted by escape probability, others are not.
Peter Woitke, Jan. 2025