Chemical rates¶
Databases¶
ProDiMo computes the gas and ice chemical content at each grid point. It can compute the chemical abundances in steady-state by default or in a time-dependent way.
ProDiMo works with several chemical rate databases (mostly gas phase) such as
- UMIST, versions 2006, 2012, 2022, https://umistdatabase.uk
- KIDA, versions 2011, 2014, 2024, https://kida.astrochem-tools.org
- and OSU.
For a detailed description of typical chemical networks used with ProDiMo see Kamp et al. (2017). In addition to those chemical databases also an additional Reaction input file is required. The additional rates can be found in Reactions.in.csv (in previous not supported versions, it is called Reactions.in) If Reactions.in.csv is not present in the model directory (1st try path) it will be read from data/ChemicalNetwork. The addition reaction files is a compilation of rates from various sources (e.g. neutral-neutral reaction rates from the NIST database, see below).
Although ProDiMo v3.0 uses a modified UMIST2012.dat rates by default, there is no endorsement of this particular network. There can be substantial differences in the rate coefficient values, in the number of reactions, or in branching ratios, ... The differences can result in for example large variation in line fluxes.
When a model is run, an output file with all the reactions and their rates is written (Reactions.out). The Reactions.out file contains all the reactions accounted for in the particular model.
The chemical network has a strong influence on the output from ProDiMo. It is advisable to describe with enough details the exact network(s) used for your model(s) in the papers/reports/thesis.
Please always cite the chemical databases correctly in your ProDiMo paper. Details for the proper citation are given below.
The default chemistry in ProDiMo is a combination of photodissociation region, dense molecular cloud, and warm dense gas chemistry. It includes:
- photo-reactions: photoionization, photodissociation
- cosmic-ray/stellar particle induced chemistry
- gas-phase reactions: 2-body and a few 3-body reactions, a few state-specific reactions (excited H2)
- freeze-out/desorption on dust grains. Desorption can be thermal, cosmic-ray induced, and photodesorption
- PAH charging and freeze-out (single size PAH)
- deuterium chemistry if deuterated species are present in the Species.in file
It does not include by default (i.e. specific flags have to be enable to account for the following chemical processes):
- warm and cold surface chemistry (apart from the H2 formation on dust grains from Cazaux & Tielens 2002; 2004; and 2010) with the corrected energies
- Xray chemistry
- Grain charge chemistry
A discussion on chemical networks can be found:
- Kamp et al. 2017
- Thi 2015 for a lecture note
- Kanwar et al. 2024 for hydrocarbon chemistry
- Thi et al. 2020, Thi et al. 2020b, Rocha et al. 2023 for the surface chemistry treatment in ProDiMo
Temperature range for the chemical rates¶
TODO: move this to another place (implementation specific stuff)
All rates in the networks are given with a range of validity. By default ProDiMo will use the rate coefficients and the appropriate rate formula to extrapolate to temperatures outside the range of validity. If one wants to cap the rates to the values at then end of the valid temperature range, please state in Parameter.in
.false. ! TextrapolateIn addition, a few rates (gas-phase) are provided with different rate coefficients for different temperatures ranges. The appropriate rate will be used given the gas temperature. When there is no extrapolation the rate using the extreme rate coefficients are used.
TODO: move the how to switch databases etc. to another place. Here just the data should be described.
UMIST¶
UMIST 2012¶
By default from v3.0, ProDiMo uses an augmented UMIST 2012 database McElroy et al. (2013). The database file can be found here UMIST2012.dat. The default UMIST2012.dat database file also includes the collider reactions (i.e. is not identical to the version from the UMIST webpage) from UMIST2006.dat and rates that involve tunnelling at low temperatures from Meisner et al. 2019. The rates with tunnelling effects are much higher for low temperature gases.
An error in the original UMIST2012 rate file has been corrected: In the original rate file (original rate UMIST2012_NOCL.dat)
928,CR,He,CRPHOT,,He+,e-,,,1.30e-17,0.00,0.2,L,10,41000,C,"OSU09""06_CRPHOT.notes"In the corrected UMIST2012 rate file with the rate refactor has been set to 1e-60:
928,CR,He,CRPHOT,,He+,e-,,,1.00e-60,0.00,0.2,L,10,41000,C,"OSU09""06_CRPHOT.notes""FIXED because of ionization potential"The reaction does not exist any more in the UMIST2022*.dat file rates (see bellow).
If you want to use the file without collider reactions and without Meisner rates, you can use UMIST2012_NOCL.dat. See bellow how to use an alternative UMIST-format network file. The absence of collider reactions + Meisner low-temperature rates and the presence of the incorrect reaction may strongly impact the chemistry and result in significant differences in the output when using UMIST2012.dat and UMIST2012_NOCL.dat.
UMIST 2006¶
The UMIST2006 network can be still used (Woodall et al. 2007). The database file is included in the ProDiMo data directory (file UMIST2006.dat). This can simply be done by setting the switch:
.true. ! UMIST2006However, we recommend using the more recent corrected version from 2012 even with older versions of ProDiMo. Notice that the UMIST2006.dat network does not have the incorrect He + CRPHOT reaction.
Additionally one can also provide an alternative database (has to be in UMIST format) by using
yourdatabase.dat ! UMISTDBThen the rates are read from the file yourdatabase.dat which has to be in the ProDiMo_datapath directory or better in your local model directory (see Adapt chemical data files). In case one uses a custom database please check the reaction types available in ProDiMo (see chemistry_solver_design).
For other changes/fixes to the original UMIST database file, please check the git history.
The .true. UMIST2012 flag without the ~ ! UMISTDB flag enables the UMIST2012 base network + the collider reactions from the UMIST2006 network that were removed from the UMIST2012 base network.
Other collider reactions are present in Reactions.in.csv especially the three-body H2 formation H + H + M (see bellow).
UMIST 2022¶
22/03/2024 and ProDiMo > v3.0
The UMIST 2022 rates (Millar et al. 2024) with and without the collider reactions added are available from data/ChemicalNetwork/UMIST2022 (master) as UMIST2022.dat. Note that the naming convention is UMIST2012, i.e. older species files are valid.
The files are UMIST2022.dat (only the UMIST 2022) and UMIST2022_CL.dat (with the collider reactions from UMIST 2006 added). The extra tunneling rates from Meisner et al. (2019) have been added to UMIST2022_CL_Meisner.dat in the same directory.
Please cite both papers if these rates are used.
The rate files have been created from the original rate file rate22_final.rates.csv downloaded here The new naming in rate22 has been converted back to the 2012 naming convention (see process_umist2022.py for more and Table 2 of the paper).
FIXME: where it this process ... file
The Meisner et al. rates add the rates in a temperature range (lower temperature) not covered by the default UMIST 2022 rates. Those tunnelling rates are important to form species at low gas temperatures.
| file | rates included |
|---|---|
| UMIST2022.dat | default UMIST 2022 with corrections |
| UMIST2022_CL.dat | above + CL |
| UMIST2022_CL_Meisner.dat | above + Meisner et al. |
For a standard user, in Parameter.in, set to use the UMIST2022_CL_Meisner.dat network
.true. ! UMIST2022Alternatively for an advanced user:
FIXME: if one does not changes those files copying is not necessary
- copy this file to the model directory
-
in Parameter.in
.true. ! UMIST2022 UMIST2022/UMIST2022.dat ! UMISTDBor
.true. ! UMIST2022 UMIST2022/UMIST2022_CL.dat ! UMISTDBor
.true. ! UMIST2022 UMIST2022/UMIST2022_CL_Meisner.dat ! UMISTDB
This makes the use of the UMIST2022.dat, UMIST2022_CL.dat or UMIST2022_CL_Meisner.dat explicit.
FIXME: We actually used UMIST2006 naming I think ... also this is mentione several times. UMIST 2012 and 2022 have different list of species. Here is the official list for UMIST2022 rate22_revised_CtoO_0.44.specs. Please use the UMIST2012 naming.
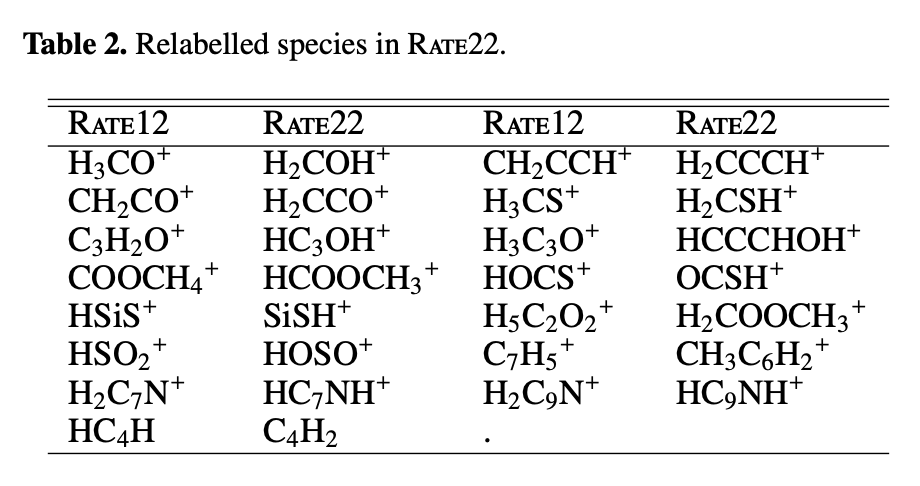
More information is available in the file UMIST2022_readme_notes.txt in the /data/ChemicalNetwork/UMIST2022/ directory.
FIXME: one should also say which species file, that defines e.g. the number of rections etc.
Here is the summary for large DIANA standard chemical network (UMIST2016 + ProDiMo extra rates vs UMIST2022 + ProDiMo extra rates):
UMIST2012: …………………
INIT_REACTIONS: ...
Init chemical Reactions ...
read: ChemicalNetwork/UMIST2012_Meisner.dat
no of reactions with multiple T-fits: 25
having included 2330 chemical reactions
Add chemical Reactions ...
read: ChemicalNetwork/Reactions.in.csv
no of reactions with multiple T-fits: 0
total 3046 chemical reactions (716 from Reactions.in.csv)
UMIST2022: ……………………
INIT_REACTIONS: ...
Init chemical Reactions ...
read: ChemicalNetwork/UMIST2022/UMIST2022_CL_Meisner.dat
no of reactions with multiple T-fits: 49
having included 2420 chemical reactions
Add chemical Reactions ...
read: ChemicalNetwork/Reactions.in.csv
no of reactions with multiple T-fits: 0
total 3115 chemical reactions (695 from Reactions.in.csv)
So we have now 69 reactions more total:
48 new reactions
21 reactions are now taken from UMIST2022 instead of Reactions.in.csvKIDA gas phase network¶
The KIDA rate database for astrochemical modelling has been compiled by Valentine Wakelam and co-workers (See KIDA website).
FIXME: Again how to change them should be somewhere else.
The switch:
.true. ! KIDA_rateswill make ProDiMo to use the high-temperature KIDA network (file KIDA_ProDiMo_2011.dat) instead of the UMIST 2006/2012 database. The additional rates in Reactions.in will be treated the same way as before according to the switches.
To use the 2014 version of KIDA (file KIDA_ProDiMo_2014.dat) you have to additionally set
KIDA_ProDiMo_2014.dat ! KIDADBFIXME: this is a general statement not just for KIDA put it more prominently
One can also use a different file as long as it is in the KIDA format and does not include any Reaction types that are unknown by ProDiMo (see chemistry_solver_design).
KIDA 2022¶
KIDA_ProDiMo_2022.dat ! KIDADBKIDA 2024 (this requires the master version)¶
gas_reactions_kida.uva.2024.in ! KIDADBThe file has not been altered from the KIDA website
The KIDA website hosts various networks used in papers. ProDiMo does not support those networks. The ProDiMo developers can implement those networks on a collaboration basis.
Citation
- KIDA 2011 release: Wakelam+ (2012)
- KIDA 2014 release: Wakelam+ (2015)
- KIDA 2022 release: Clement+ (2023) FIXME: is this really officially KIDA 2022?
- KIDA 2024 release: Wakelam+ (2024)
For details on the citation of the KIDA databse please check the KIDA website credits.
OSU (Ohio State University)¶
The OSU rate database for astrochemical modelling has been compiled by Eric Herbst and co-workers. The OSU database has been discontinued. KIDA is a follow-up database of OSU.
The switch:
.true. ! OSU_rateswill make ProDiMo use the high-temperature OSU network (files osu_09_2012_ht_ProDiMo.dat and osu_pol_data.dat) instead of the UMIST 2006 database. The additional rates in Reactions.in will be treated the same way as before according to the switches. Those rates are appropriate for warm gas chemistry.
Citation
- A New Network for Higher-temperature Gas-phase Chemistry. I. A Preliminary Study of Accretion Disks in Active Galactic Nuclei*, Harada, Nanase; Herbst, Eric; Wakelam, Valentine, 2010, ApJ. 721, 1570H and the Erratum
OSU/AlCHEMIC¶
Dimitry Semenov added surface reactions based on the Hasegawa & Herbst 1995 network to the standard OSU gas-phase network. The main purpose of the Alchemic network is to enable a comparison between parts of the ProDiMo surface chemistry implementations (see /examples/Benchmarks/Benchmark_SURFACE_CHEMISTRY). Not all the surface chemistry physics that are in ProDiMo is tested by the benchmark.
.true. ! OSU_Alchemic : use the network of the benchmark
.false. ! other_reactions
.true. ! surface_chemistryIf you set the
.true. ! other_reactionsthen reactions in the Reactions.in file are considered as well according to the switch used (only_add, replace)
Citation
- Chemistry in disks. IV. Benchmarking gas-grain chemical models with surface reactions*, Semenov et al. A&A, Volume 522, id.A42, 12 pp. 2010
Multiple network statements in Parameter.in¶
FIXME: Again there should be a dedicated page on how to switch databases One should avoid conflicting network assignments.
STAND2020 network¶
Since April 2023, you can also add reactions from the STAND2020 network for planetary atmospheres, including lots of three-body reactions (Rimmer & Helling 2016, ApJSS 224, 33; Rimmer et al. 2021, Planetary Science Journal 2, 25). This is different than for UMIST/KIDA/OSU/ALCHEMIC, which are alternative networks that cannot be merged together.
The user can choose to between the database via simple switches in Parameter.in, for example
FIXME: this example will not work as is because of the conflict between UMIST and KIDA (confusing for th e user)
.true. ! UMIST2012
onlyadd ! handle_UMIST (erase/overwrite/onlyadd)
.true. ! KIDA_rates
KIDA_ProDiMo_2014.dat ! KIDADB
.true. ! STAND2020Choose one of UMIST or KIDA, not both. You can add the STAND rates to both.
In addition extra reactions in Reactions.in.csv or Reactions.in are also add (depending on the flag used).
ProDiMo extra reaction network¶
The standard networks do not provide all the required reaction rates to model a protoplanetary disk environment. In addition to the base chemical database, ProDiMo will use the reactions and rates in the file Reactions.in.csv. This file is located at /data/ChemicalNetwork. The format is the same than the UMIST2012.dat or UMIST2022.dat file networks.
The Reactions.in.csv may contain duplicate reactions with the base database. How those duplicate rates are accounted for depending on the flag. It is recommended to use in Parameter.in
onlyadd ! handle_UMIST : handle UMIST-data (erase/overwrite/onlyadd)This flag/switch means that the UMIST rates (it also works for other base networks) will be used. Only reactions involving species in Species.in that do not exist in the base network will be added to the list of reactions. The Reactions.in.csv is a collection of rates from various sources:
- The H2 formation on dust grain (even without explicit surface chemistry)
- Photodissociation code network, e.g. Tielens and Hollenbach 1985 (TH85), Meudon PDR code
- Xray reactions involving doubly ionised species: Fe++, Mg++, ...
- Reactions with vibrationally excited H2 (called H2exc). The reactions are neutral-neutral reactions involving H2 and with an activation barrier. The H2 v=1 level energy o is subtracted to the energy of the activation (third numerical term) in the non-excited H2 rate.
- Adsorption and Desorption reactions (photo induced and cosmic-ray induced desorption)
- Surface chemistry network based on rates from Hasegawa and Herbst 1992 (HH92) with updates (most on the activation energy)
- Warm surface chemistry rates to form H2, HD and phyllosilicate
- Deuterium chemistry to model HD, DCO+, ...
- Charge exchange reactions in the gas-phase and with charges on dust grains
- PAH charging, freeze-out, hydrogenation and de-hydrogenation
- H2 formation on PAHs
- other types of reactions in specific papers
The sources for the reactions are diverse (TH85, NIST, HH92, Garrod, STAND, ...).
If you want to read the file in csv format, you need to use
.true. ! read_reactions_in_csvin which case Reactions.in.csv is read from the directory /data/ChemicalNetwork/.
You can also provide your own (with the correct format) extra network with it own filename using
yourreactions.in.csv ! reaction_file For example,
.true. ! read_reactions_in_csv
Reactions_galaxies.in.csv ! reaction_fileThe Reactions.in.csv file follows the formatting of the UMIST2012 database. As such, reaction types, gas-phase T/F flags are now explicitly included as well as temperature ranges for the rates (T_min, T_max, for now set to dummy values). This means that the user has to ensure that these reaction types, flags are correctly set.
If the final networks contain a very large number of reactions, one needs to make some change to the code itself. You should contact one of the developers.
Photorates¶
The photorates are taken from the database maintained by Ewine van Dishoeck at https://home.strw.leidenuniv.nl/~ewine/photo/. If the rates are recent (<5 years), please cite the original papers.
TODO: Do we use data fom these databases? Other databases: The Southwest Research Institute
DEAD LINK http://phidrates.space.swri.eduprovides high-resolution cross-sections. and the Mainz group atDEAD LINK http://www.atmosphere.mpg.de/enid/2295